
ESPE2016 Poster Presentations Gonads & DSD P1 (48 abstracts)
Genotyping Patients with Differences of Sex Development: 25 Years of Investigation of an Italian Population of 308 Cases (194 46,XY and 114 46,XX)
Lilia Baldazzi a , Soara Menabò b , Federico Baronio a , Rita Ortolano b , Alessandra Cassio b , Laura Mazzanti b & Antonio Balsamo b
aS.Orsola-Malpighi University Hospital, Bologna, Italy; bDepartment of Medical and Surgical Sciences University of Bologna, Bologna, Italy
Background: Differences of sex development (DSDs) (conditions with atypical development of chromosomal, gonadal or anatomic sex) are classified into three groups: sex chromosome DSD, 46,XYDSD and 46,XX DSD. Around 1 newborn in 5000 presents ambiguous genitalia with a major challenge for male or female assignment. The identification of a genetic cause can contribute to a correct diagnosis and to optimize both management and genetic counselling.
Objective and hypotheses: To describe the results of the diagnostic activity on a large cohort of cases (chromosomal DSD excluded), mostly from the Nord-Est Italian regions referring to our centre in the period 1991–2016.
Method: Hundred and ninety-four cases with 46, XY DSD and 114 cases of 46,XX DSD where analysed by Sanger sequencing and/or MLPA for the major candidate genes/regions for the specific DSD condition.
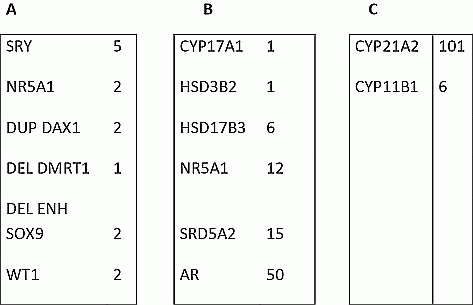
Results 46, XY DSD: A genetic cause was identified in 14 out of the 27 Gonadal dysgenesis (A); in 85 out of the 133 defects of androgen synthesis or action (B); in 11 (9 AMH) out of 14 PMDS; in 1 (MAMLD1) out of 20 isolated ipospadias. 46, XX DSD: a genetic cause was identified in 4 (SRY+) out of 7 testicular DSD; in 100% of 107 virilized females with androgen excess (C).
Conclusion: The mutation detection is: 57.2% in 46, XY DSD (111/194) with gonadal dysgenesis and isolated hypospadias as the less characterized; 93.9% (107/114) in 46, XX DSD, confirming both the large prevalence of defects of adrenal steroidogenesis and the lack ok knowledge for testicular DSD. A panel of a limited n. of genes is sufficient to obtain a good mutation detection for group B.
}
Article tools
My recent searches
My recently viewed abstracts

ESPE Abstracts
© Bioscientifica 2025 |
Privacy policy |
Cookie settings
BiosciAbstracts

Bioscientifica Abstracts is the gateway to a series of products that provide a permanent, citable record of abstracts for biomedical and life science conferences.
Find out more