
ESPE2016 Rapid Free Communications Growth : Mechanisms (8 abstracts)
Whole Exome Sequencing Identifies a GH1 Gene Mutation Causing Familial Isolated Growth Hormone Deficiency with Normal Peak Growth Hormone Concentrations
Catalina Cabrera Salcedo , Vivian Hwa , Leah Tyzinski , Melissa Andrew , Philippe Backeljauw & Andrew Dauber
Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA
Background: Familial isolated growth hormone deficiency (IGHD) type II is autosomal dominantly inherited and caused by splice-site mutations and nucleotide substitutions in the GH1 gene. The missense mutation R183H is a well-described genetic variant that causes familial IGHD type II. Individuals with this mutation have releasable GH stores, but GH secretion is severely reduced resulting in short stature.
Objective and hypotheses: This study aimed to report two female patients from a four-generation family with 6 individuals with short stature who carry the R183H mutation in GH1.
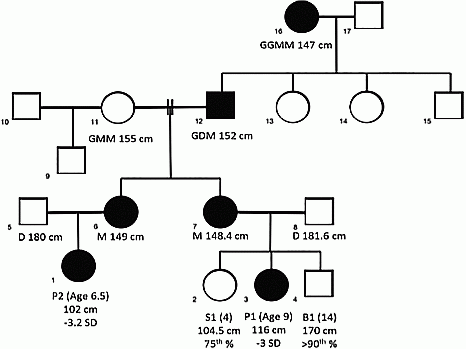
Method: Two first degree female cousins with significant short stature were evaluated in the Cincinnati Center for Growth Disorders. Patient 1 (P1) was 9 years old and her height was 116 cm (−3.0 S.D.). She had been previously diagnosed with ISS based on a normal GH stimulation test. Patient 2 (P2) was 6.5 years of age and her height was 102 cm (−3.2 S.D.) After extensive evaluation whole exome sequencing (WES) was performed.
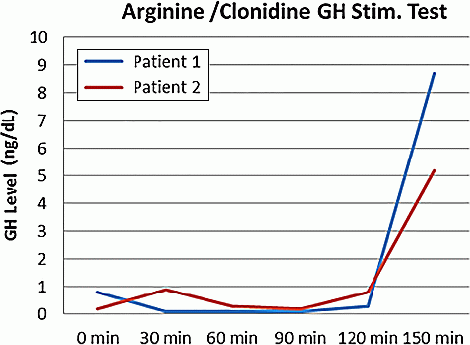
Results: Via WES, we identified the pathogenic variant R183H in GH1 which segregated in the affected individuals. P1 and P2 underwent GH stimulation tests and were found to have delayed peak GH responses. P1 had a peak GH level of 8.7 ng/dl which is considered normal, and P2 had a peak GH level of 5.2 ng/dl.
Conclusion: Patients with IGHD type II secondary to the R183H GH1 mutation can exhibit variable peak GH responses. Genetic testing should be strongly considered in cases of familial short stature even when peak stimulated growth hormone concentrations are normal. It remains unclear whether or not adult patients with this mutation suffer the consequences of adult GH deficiency. We are implementing a protocol to investigate body composition, skeletal integrity, cardiovascular risk profile and the overall quality of life in the affected adults of this kindred.
Article tools
My recent searches
My recently viewed abstracts

ESPE Abstracts
© Bioscientifica 2024 |
Privacy policy |
Cookie settings
BiosciAbstracts

Bioscientifica Abstracts is the gateway to a series of products that provide a permanent, citable record of abstracts for biomedical and life science conferences.
Find out more