
ESPE2015 Poster Category 3 Adrenals (47 abstracts)
Deletion Mapping in Xp21 for a Family with Complex Glycerol Kinase Deficiency Using Array-Based Comparative Genomic Hybridisation
Zhangqian Zheng a , Feihong Luo a , Bingbing Wu b & Miaoying Zhang a
aDepartment of Endocrine and Inherited Metabolic Diseases, Children’s Hospital of Fudan University, Shanghai, China; bPediatric Research Institute, Children’s Hospital of Fudan University, Shanghai, China
Aims: Complex glycerol kinase deficiency is caused by partial deletion of Xp21, which includes the genes responsible for glycerol kinase deficiency, adrenal hypoplasia congenita, Duchenne muscular dystrophy and intellectual disability. There are no definite dysmorphic features for this syndrome. The diagnosis is based on clinical and laboratory findings. Usually the first and most severe are the signs of adrenal hypoplasia, which, if not cured, may lead to death in a short time. The symptoms of glycerol kinase deficiency occur also early in life, but they may be masked by the deficiency of mineralocorticoids. Duchenne muscular dystrophy appears in childhood and is always accompanied by certain symptoms.
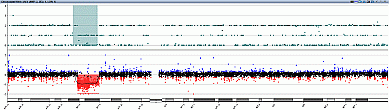
Methods: Genomic DNA from the proband and his parents were extracted from peripheral blood leukocyte. Array-based comparative genomic hybridization of DNA from the family’s peripheral blood lymphocytes was performed.
Results: The proband, a male neonate, is the first child of healthy nonconsanguineous Chinese parents. He was born by uterine-incision delivery after 41 weeks of gestation. His birth weight was 4.0 kg. After 22 days of born, he was admitted with hyperbilirubinemia. He was diagnosed of temporary hypothyroidism, congenital adrenal hypoplasia, pneumonia and liver function damage. Treated with antibiotics and rehydration, he recovered. One month later, he presented with feeding problem and lethargy and was admitted again. He was suffered with adrenal hormone insufficiency, hypertriglyceridemia and highly increased creatine kinase. We performed array CGH and confirmed the deletion region of Xp21. Then we checked his parents and found his mother was the carrier and his father was normal. Proband’s aCGH result: chrX: 28,612,178-37,392,685. The same deletion in Chromsome X was found in mother.
Conclusion: A deletion of the Xp21 chromosomal region is found in both mother and the child. Before decades, complex glycerol kinase deficiency was usually diagnosed by clinical manifestation and Many of these patient deletions had been mapped by PCR and their breakpoints confirmed by sequencing. We confirmed that array CGH analysis can be performed to confirmed the diagnosis of patients along with the biochemical exams.
}
Article tools
My recent searches
My recently viewed abstracts

ESPE Abstracts
© Bioscientifica 2026 |
Privacy policy |
Cookie settings
BiosciAbstracts

Bioscientifica Abstracts is the gateway to a series of products that provide a permanent, citable record of abstracts for biomedical and life science conferences.
Find out more