
ESPE2016 Poster Presentations Syndromes: Mechanisms and Management P1 (36 abstracts)
A Case of Patient with Rubinstein-Taybi Syndrome Type 2 with Complete Deletion of EP300 Gene and Complex Phenotype
Elisa Santoro a , Romana Marini a , Antonio Novelli b , Viola Alesi b , Maria Lisa Dentici c & Marco Cappa a
aBambino Gesù Children’s Hospital – IRCCS; Department of Endocrinology and Diabetes, Rome, Italy; bBambino Gesù Children’s Hospital – IRCCS; Laboratory of Medical Genetics, Rome, Italy; cBambino Gesù Children’s Hospital – IRCCS; Medical Genetics Unit, Rome, Italy
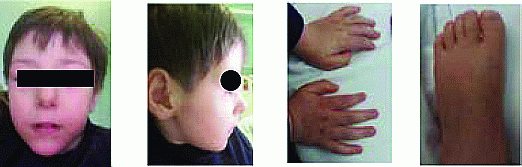
Background: Rubinstein-Taybi syndrome (RSTS) is a rare genetic syndrome characterized by postnatal growth retardation, intellectual disability, microcephaly, peculiar facial features, broad thumbs and big toes and other organs malformations. There are two forms: RSTS type 1 characterized by CREBBP gene mutations (16p13.3); RSTS type 2 dues to mutations/ deletions in EP300 gene (22q13.2). The type 2 is associated with mild phenotype with possible absence of the typical diagnostic signs as broad thumbs and big toes.
Clinical case: V.B.G has come to our attention at the age of 4 years for diagnostic study. He was born at 36 weeks of gestational age by caesarean section for IUGR. At birth he presented congenital heart disease, hypospadias, post-axial appendix of left hand, choroidal cysts, dysmorphia of corpus callosum, rostrum hypoplasia, agenesis of right olfactory bulb, enlargement of cerebral ventricles and CSF spaces. The array-CGH showed 22q13.2 deletion of 1.69 Mb with diagnosis of RSTS type 2. At the age of 3 years he presented psychomotor regression with onset of focal seizures. At our observation the child had growth retardation, dysphagia for liquids, constipation, characteristic face, broad toes, limbs dystonia, absent language.
Conclusion: In literature there are known 34 cases of RSTS associated with EP300 alteration. Of these, only one has complete gene deletion (del22q13.2) of 376 Kb. This patient has a complex phenotype in contrast with “mild” phenotype of the other patients. Because of the larger size of our child’s deletion his clinical complexity could also be related to the involvement of adjacent genes. In addition it would be useful scanning EP300 gene also on other tissues to identify a possible mosaicism. Our case represents the second known patient suffering from type 2 RSTS with complete EP300 deletion and clinical features in contrast with a “mild” phenotype. A genotype-phenotype correlation for EP300 disruptive mutations is needed.
}
Article tools
My recent searches
My recently viewed abstracts

ESPE Abstracts
© Bioscientifica 2026 |
Privacy policy |
Cookie settings
BiosciAbstracts

Bioscientifica Abstracts is the gateway to a series of products that provide a permanent, citable record of abstracts for biomedical and life science conferences.
Find out more